
HOME / Departments / Chemistry / Theoretical Chemistry
Theoretical Chemistry
-
- NAKANO Haruyuki, Professor
- WATANABE Hiroshi, Associate Professor
- WATANABE Yoshihiro, Associate Professor (Lecturer)
- SUZUKI Satoshi, Assistant Professor
- KAWASHIMA Kyohei, Assistant Professor
- Yang Chen, Assistant Professor
- Researches of our group is devoted to the theoretical chemistry based on the first principles of quantum and statistical mechanics. Our main research objective is to develop theoretical methods for electronic structures, properties, and chemical reactions of molecules, clusters, and molecular assemblies. Our focus is especially on the electron correlation theory for molecular systems, molecular theory of solvation, and relativistic molecular orbital theory. We develop mathematical methods, algorithms, and computer software, and approach various interesting chemical phenomena such as solvatochromism, co-solvent effects, and pKa of drug molecules.
1. Electron correlation theory for molecular systems—Multireference perturbation theory
Multireference perturbation theory (MRPT) based on multiconfiguration (MC) reference functions has become a basic and practical tool for studying the electronic structures of molecules and the potential energy surfaces of chemical reactions. MRPT takes account of both static and dynamic electron correla-tions and thus can obtain accurate relative energies, including reaction, activation, and excitation energies, within a chemical accuracy (i.e., a few kcal/mol). We have developed an MRPT using MC functions that we call “multiconfigurational quasidegenerate PT (MC-QDPT)”. It is a multiconfiguration basis multireference-state method based on van Vleck PT and includes multireference Møller–Plesset (MRMP) PT, a single-reference-state method based on Rayleigh–Schrödinger PT, as a special case. In particular, a recent version of MC-QDPT uses general multiconfiguration reference functions (GMC-QDPT or GMC-PT). GMC-QDPT imposes no restriction on the reference space, so it is much more compact than complete active space (CAS)-based MRPT.
H. Nakano, J. Chem. Phys. 99,7983 (1993); H. Nakano, R. Uchiyama, K. Hirao, J. Comput. Chem. 23, 1166 (2002)
2. Molecular theory of solvation—Hybrid theory of quantum mechanics and statistical mechanics
Solvent effect on the electronic structure of molecules has attracted a lot of attention in the fields of chemistry, physics, and biophysics, because many chemical reactions occur in solution. An electronic structure in solution is affected by surrounding solvent molecules which have large degrees of freedom. Therefore, the solvent environment should be treated by statistical mecanics. We have developed hybrid theories by combining quantum mechanics and statistical mechanics for the electronic structure of solvated molecules such as molecular Ornstein-Zernike self-consistent field (MOZ-SCF), quantum mechanics/molecular mechanics reference interaction site model (QM/MM/RISM), and the fragment molecular orbital/ three-dimensional reference interaction site model (FMO/3D-RISM) theory. These methods are available for variety of chemical and biological processes in solution that involve electron transfer, molecular recognition, photochemical reaction, and so forth.
N. Yoshida, S. Kato, J. Chem. Phys. 113, 4974 (2000); N. Yoshida, et al., J. Mol. Liquids. 159, 83 (2011); N. Yoshida, J. Chem. Phys. 140, 214118 (2004)
3. Relativistic molecular orbital theory
3.1 Four-component multireference PT
The importance of simultaneous consideration of relativistic and electron correlation effects for describing the electronic structures and chemical reactions of systems involving heavy atoms is now well-recognized. Many methods for describing electronic structures, including the electron correlation effect, have been transferred to the four-component relativistic level: Møller–Plesset (MP) perturbation, configuration interaction (CI), coupled-cluster (CC) methods based on the Dirac–Hartree–Fock (DHF) wave function, and the Dirac–Kohn–Sham method. Multireference (MR) CI and CC methods are also available through relativistic program packages. However, as in the non-relativistic case, the MRCI and MRCC methods require much computational cost. Lower cost multireference methods are needed. We have extended our GMC-QDPT to a relativistic version with four-component general MC reference functions.
M. Miyajima, Y. Watanabe, H. Nakano, J. Chem. Phys. 124, 124302 (2006); R. Ebisuzaki, Y. Watanabe, Y. Kawashima, H. Nakano, J. Chem. Theory Comp. 7, 998 (2011)
3.2 Necessity of including the negative energy space
The no virtual pair approximation (NVPA), which do not consider the electron excitation to the so-called Dirac sea, is widely accepted in electronic structure calculation of atoms and molecules using four-component relativistic methods. The effect of the electron excitation to the Dirac sea, the contribution from the negative energy space, was expected to be relatively small. We recently performed a critical assessment on the effect from the negative energy space in four-component relativistic electronic structure calculations, and found that the inclusion of the negative energy space is essential for accurate or exact calculations. We are currently investigating the physical interpretation of the NVPA and the contribution from the negative energy space in four-component relativistic electronic structure calculations.
Y. Watanabe, H. Nakano, H. Tatewaki, J. Chem. Phys. 126, 174105 (2007); Y. Watanabe, H. Nakano, H. Tatewaki, J. Chem. Phys. 132, 124105 (2010); H. Tatewaki, Y. Watanabe, Chem. Phys. 389, 58 (2011)
4. Some recent applications
4.1 Solvent and substituent effects on the absorption spectra of Brooker's merocyanine
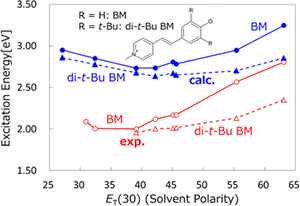
Solvatochromism is a phenomenon involving a change in the color of a solution depending on the solvent. This phenomenon is now utilized in many fields of chemistry and biology to study the bulk and local polarity in macrosystems. In the solvatochromism of BM, substituents are known to play an important role. We investigated the solvent and substituent effects on the absorption spectra of BM using the three-dimensional RISM-SCF (3D-RISM-SCF) method.
Y. Tanaka, N. Yoshida, H. Nakano, J. Comput. Chem. 36, 1655 (2015)
4.2 Co-Solvent Effect on the Proton Transfer Reaction of Glycine in a Water-Acetonitrile Mixture
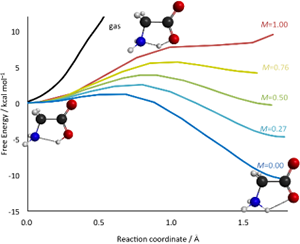
The co-solvent effect on the proton transfer reaction of glycine in a water–acetonitrile mixture was examined using the reference interaction-site model self-consistent field (RISM-SCF) theory. The free energy profiles of the proton transfer reaction of glycine between the carboxyl oxygen and amino nitrogen were computed in a water–acetonitrile mixture solvent at various molar fractions.
The detailed analysis of the co-solvent effect on the proton transfer reaction should be indispensable for a deeper understanding of the solvation in heterogeneous environments.
Y. Kasai, N. Yoshida, H. Nakano, J. Chem. Phys. 142, 204103 (2015)
4.3 Changes in pKa of drug molecule by complex formation
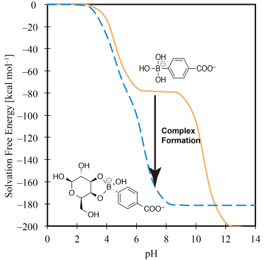
The pKa of drug molecules under the physiological conditions in the human body is a crucial issue in pharmacology, because a pKa is in close relation with solubility. The pKa change of drug molecule upon complex formation are considered by using 3D-RISM-SCF method. The result shows good agreement with experimental observation and it revealed the detail of the mechanism of pKa change.
Y. Seno, N. Yoshida, H. Nakano, J. Mol. Liquids, in Press (2016)