
HOME / Departments / Chemistry / Molecular Catalysis Chemistry
Molecular Catalysis Chemistry I
-
- KUWANO Ryoichi, Professor
- We are developing novel and/or stereoselective organic reactions by using transition-metal complexes as catalysts. The new molecular transformations may lead to the discovery and creation of new materials and medicines, which may contribute the innovative progress of technology. We have realized on 1) the highly enantioselective hydrogenation of various heterocyclic and carbocyclic arenes, 2) the palladium-catalyzed substitution of benzylic carboxylates and carbonates with various nucleophiles, 3) the palladium catalyzed [4 + 2] cycloadditions of o-(silylmethyl)benzyl carbonates, 4) the rhodium-catalyzed reactions of alkenyl acetates, and 5) the nickel- or palladium-catalyzed oxidative β-aminations of ketones and related compounds. Furthermore, we are studying on the reaction mechanism of the above catalytic reactions.
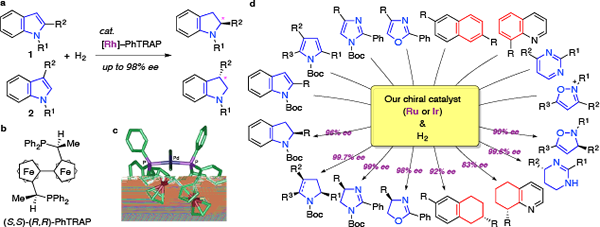
I-1. Catalytic asymmetric hydrogenation of heterocyclic and carbocyclic arenes
Nowadays, catalytic asymmetric hydrogenations of alkenes, ketones, and ketimines provide a powerful tool for preparing optically active compounds. Arenes are also attractive substrates for the asymmetric hydrogenation. However, the asymmetric reduction of arenes is a formidable target in organic chemistry.
In 2000, we reported the highly enantioselective hydrogenation of indoles 1 with a chiral rhodium catalyst, [Rh(nbd)2]SbF6–(S,S)-(R,R)-PhTRAP. The chiral catalyst produced the desired chiral indolines with up to 98% ee. This was the first success in the catalytic asymmetric hydrogenation of heteroarenes.
Since then, we have achieved high enantioselectivities for the hydrogenation of various heteroarenes, pyrroles, imidazoles, oxazoles, pyrimidines, and isoxazoles. The chiral catalyst, PhTRAP–ruthenium complex, is useful for the asymmetric hydrogenations of naphthalenes and quinoline carbocycles. Furthermore, we are challenging to the catalytic asymmetric hydrogenation of benzene rings.
I-2. Palladium-catalyzed substitution of benzylic esters with various nucleophiles
Tsuji–Trost reaction, allylic substitution of allyl esters, is a useful catalytic reaction for organic synthesis. We proved the similar benzylic substitution to proceed by a palladium catalyst. The palladium catalyst allows the benzylic carbonates and carboxylates to react with stabilized carbanions, amines, phenols, sulfinates, organoboron and organostannane compounds.

I-3. Palladium-catalyzed [4 + 2] cycloadditions ofo-(silylmethyl)benzylic carbonates
o-Quinodimethane is a useful reactive intermediate for organic synthesis. However, the intermediate is so unstable that its reaction can be controlled in stereochemistry as well as regioselectivity. We designed o-(silylmethyl)benzylic esters as a surrogate of the o-quinodimethane.
We found that a phosphine–palladium complex worked as the catalyst for the cycloaddition of o-(silylmethyl)benzylic carbonates. The o-quinodimethane precursors reacted with C=C, C=N, C=O double bonds to give the desired [4 + 2] cycloaddition products in high yields. In some cases, the catalyst controlled the rigioselectivity of the cycloaddition.
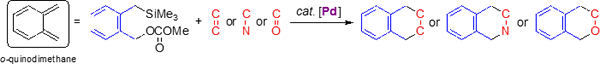
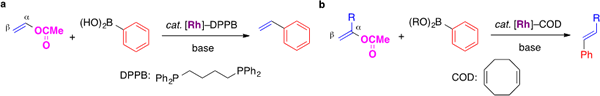
I-4. Rhodium-catalyzed reaction of alkenyl acetates
Vinyl chloride is often used for the synthesis of terminal alkenes through the cross-coupling reaction. However, the chloroalkene has serious toxicity as well as impact on ecosystem. In this project, we have developed a catalyst, which enable to use vinyl acetate as a surrogate of vinyl chloride.
We found that the Suzuki–Miyaura cross-coupling reaction of vinyl acetate with arylboronic acids proceeded in the presence of a phosphine–rhodium catalyst. In the rhodium-catalyzed reaction, the C–C bond formation took place on the α-carbon of vinyl acetate. Meanwhile, arylboronates reacted with the β-carbon of alkenyl acetates when a diene–rhodium complex was used as the catalyst.
I-5. Nickel- or palladium-catalyzed oxidative β-amination of ketones and related compounds
In the reaction of alkyl ketones, a bond is newly created on the carbonyl carbon (e.g. Grignard reaction) or a-carbon atom (e.g. aldol reaction). However, the bond formation at the β-position is an unusual reaction in classic organic chemistry.
The bond formation at the β-position was observed when a mixture of propiophenone and morpholine was heated in the presence of chlorobenzene and a nickel catalyst. The nickel-catalyzed reaction produced b-enaminone in high yield. Palladium is preferable to nickel for the β-oxidative amination of a,b-unsaturated nitriles.

Molecular Catalysis Chemistry II
-
- SUENAGA Masahiko, Lecturer
- My first research interest is a development of GUI (Graphic User Interface) for molecular modeling and computational chemistry software (such as TINKER, MSMS, Firefly, Gamess, MOPAC and Gaussian). Since 2004, I have developed GUI software, Facio, and opened it to the public as freeware, with a hope that it will be helpful to study and research on theoretical chemistry. In addition to the educational purpose, this software is indispensable to FMO (Fragment Molecular Orbital) study, since it is the only GUI for FMO-specific input and output.
My second research interest is to re-examine fundamental concepts in organic chemistry, such as steric hindrance, using NBO (Natural Bond Orbital). While MO (Molecular Orbital) views various molecular properties as a whole, NBO can view them as interaction between localized donor orbital and acceptor orbital providing a different viewpoint to organic chemistry.
II-1. Development of computational chemistry environment i.e. GUI for molecular modelling and visualization of computational chemistry calculations, as shown below
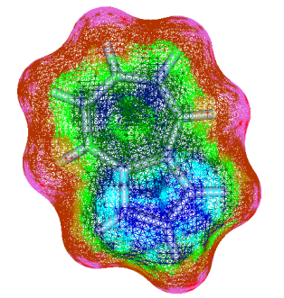

FMO (Fragment MO) is a computational method to calculate very large molecular system, such as protein, by dividing them into fragments and to evaluate interaction energy between fragments.

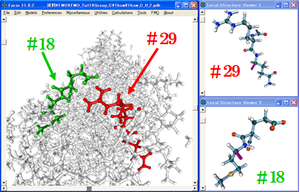

I have developed GUI (Graphic User Interface) for FMO / GAMESS and implemented in Facio. By using this GUI, one can easily make FMO fragments for a given protein or nucleic acid, generate FMO input files and visualize output files.
II-2. Theoretical study of fundamental concepts in organic chemistry with NBO
II-2.1 SOI (secondary orbital interaction) in endo rule of Diels–Alder reaction
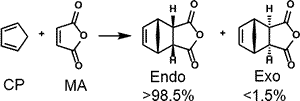
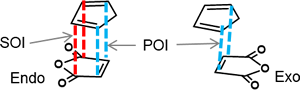
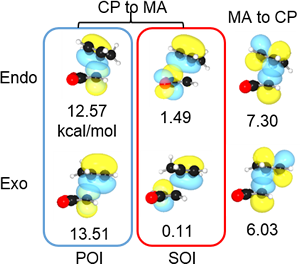
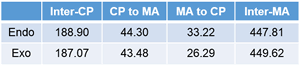
NBO interaction corresponding to the so called SOI (Secondary Orbital Interaction) is 1.49 kcal/mol, which is only 7% of total NBO interactions. In addition, there are many other interactions smaller than POI (Primary Orbital Interaction), but much greater than SOI as shown. Thus, the reason of endo rule that kinetically preferred product is endo in Diels–Alder reaction cannot be attributed solely to the so called SOI.
II-2.2 Origin of thermodynamic preference of trans-2-butene over cis-2-butene
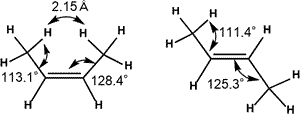
Due to steric repulsion between two methyl group in cis isomer, trans isomer is generally thought to be thermodynamically more stable.
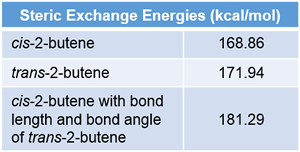
Natural Steric Analysis (in the right table) shows the steric repulsion of cis isomer seems to be released with increasing bond angle Csp2-Csp2-Csp3 (125.3° to 128.4°).
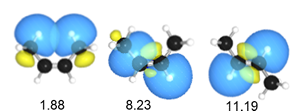
Actually, pairwise steric exchange energy between approximate hydrogen atoms of two methyl groups is 1.88 kcal/mol, which is far less than that of σ(CC)-σ(CH) (8.23) of cis isomer or σ(CH)-σ(CH) (11.19) of trans one.
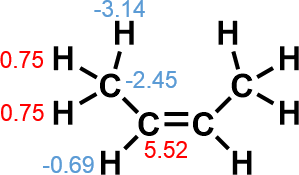
Atomic energy of approximate hydrogen atom is large negative value (−3.14), which suggest the existence of attractive force between them, rather than repulsion. On the other hand, that of central sp2 carbon is large positive value (5.52), which suggest avoidance of steric repulsion resulted in deformation of the skeleton and increase of atomic energy not in methyl carbon, but in sp2 carbon.