HOME / Departments / Chemistry / Biomolecular Chemistry
Biomolecular Chemistry
-
- NOSE Takeru†, Professor
- SUYAMA Keitaro†, Assistant Professor
- NAKAGAWA Natsumi†, Assistant Professor
- † Faculty of Arts and Science
- Biological phenomena are governed by biomolecules with diverse structures and physiological functions. A key question is whether such molecules can be rationally designed. To address this, we explore strategies for creating novel biomolecules with new structures and functions through chemical synthesis, structural analysis, activity assays, computational science, and molecular biology.
1. Structure–function relationship of elastin-derived peptides
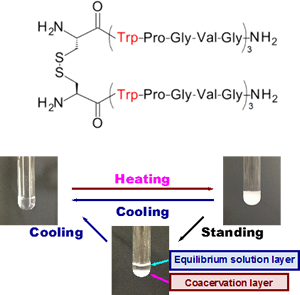
Elastin, a core protein of elastic fibers, exhibits coacervation. Coacervation is promoted by molecular interactions and shows temperature-dependent reversible association/dissociation under physiological conditions. Because of this unique characteristic, elastin and elastin-derived peptides have been considered to be useful as base materials for developing various biomedical products, skin substitutes, synthetic vascular grafts, and drug delivery systems. Although elastin-derived polypeptide (Val-Pro-Gly-Val-Gly)n has also been known to demonstrate coacervation properties, a sufficiently high (VPGVG)n repetition number (n > 40) is required for coacervation. In our recent studies, we newly developed a series of elastin-derived peptide dimers, (Cys-(Phe-Pro-Gly-Val-Gly)5)2 and (Cys-(Trp-Pro-Gly-Val-Gly)3)2, possessing a high coacervation potential. These novel dimeric peptides exhibited coacervation at significantly lower concentrations and temperatures than the commonly used elastin-derived peptide analogs; this result suggests that the coacervation ability of the peptides is enhanced by dimerization. On the basis of these findings, we are developing more potent elastin-derived peptide analogs with biological and chemical functions.
2. Determination and discovery of receptor-binding molecules
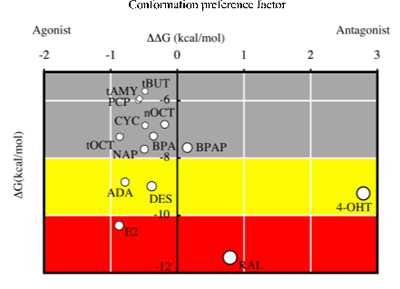
We conducted risk assessment studies on endocrine disrupting chemicals (EDCs) by using a combination of computational and biochemical techniques. Nuclear receptors (NRs) are the main target of EDCs and play a central role as transcription factors in biological processes. Most of these NRs possess a ligand-binding domain (LBD) for binding a hormone or a hormone-like chemical. If chemicals that can bind to the LBD are identified, this information would, in turn, be very helpful for identifying chemicals that can bind to NRs. Such chemicals can be considered as dangerous EDCs that cause serious disruptions in the endocrine system. We established a novel screening method to study endocrine-disrupting chemicals by means of in silico docking calculations. We used the crystal structures of both agonist-bound LBDs and antagonist-bound LBDs as templates in the docking calculations, and estimated binding energies to discriminate between agonist and antagonist binding. This agonist/antagonist differential-docking screening (AADS) method did predict, for example, 4-(1-adamantyl)phenol as an agonist of the human estrogen receptor α. The AADS method is an approach that appears to predict both the binding ability and the agonist/antagonist function of chemicals for the target nuclear receptors.
3. Rational molecular design of enzyme inhibitors
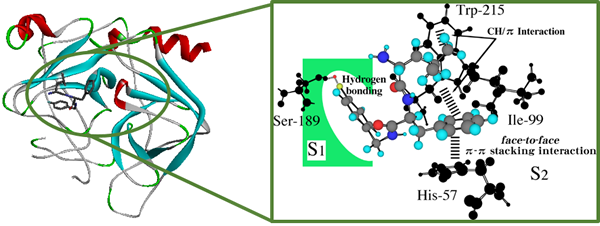
The dipeptide inhibitor D-Leu-Phe-NHBzl (pF) inhibits chymotrypsin via a π-π stacking interaction between the Phe-phenyl group of the inhibitor and His57-imidazole ring at the active site of chymotrypsin. This π interaction is essential for inhibition. In this study, we considered whether the docking calculation based on the conventional molecular mechanics method was applicable to strengthening the inhibitory activity of the dipeptide inhibitor. On the basis of the calculations, we are going to design and synthesize tripeptide inhibitors and to perform in vitro enzyme-inhibition tests.